SP-BRAIN: scalable and reliable implementations of a supervised relevance-based machine learning algorithm.
SP-BRAIN a supervised machine learning algorithm based on Big Data technologies and applied on the prediticition of DNA splicing sites has been released.
Classification of Large DNA Methylation Datasets for Identifying Cancer Drivers.
BIGBIOCL an algorithm that can apply supervised machine learning methods to datasets with hundreds of thousands of features for the extraction of alternative and equivalent classification models
has been published.
Combining EEG signal processing with supervised methods for Alzheimer's patients classification.
A new procedure for classifying EEG signals has been designed and applyed to real demented and Alzheimer's diseased patients.
A novel method and software for automatically classifying Alzheimer's disease patients by magnetic resonance imaging analysis.
A new paper, method, and software for classifying Magentic Resonance Images has been published in Computer Methods and Programs in Biomedicine.
TCGA2BED: extracting, extending, integrating, and querying The Cancer Genome Atlas.
A software tool to search and retrieve TCGA data and convert them in the structured BED format for their seamless use and integration has been released.
Emanuel Weitschek manages and participates to several bioinformatics and biomedical engineering projects.
For the software projects please refer to his data mining and analysis website available at dmb.iasi.cnr.it.
The aim of this work is the definition of an unique and global platform for effectively storing, searching and retrieving genomic data via distributed computing.
Our main duties are data extraction, conversion, and integration from The Cancer Genome Atlas (TCGA) database.

The data is processed with the aid of the Genome Query Language.
This work is supervised by Dr. Marco Masseroli and
Dr. Stefano Ceri from Politecnico di Milano.
Alzheimer's Disease electroencephalography signal processing
The aim of this work is to achieve an automatic patients classification from the Electroencephalography (EEG) signals involved in neuropathology diseases
(e.g., Alzheimer and Mild Cognitive Impairment (MCI)) in order to aid medical doctors in the right diagnosis formulation.
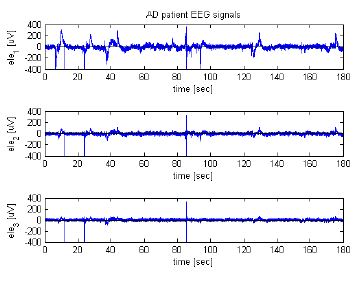
The analysis of the biological
EEG signals requires effective and efficient computer science methods to extract relevant information. Specifically, in our work, we are carrying on these
analysis steps: (i) pre-processing of EEG data; (ii) processing of the EEG-signals by the application of time-frequency transforms
(e.g., Fourier Transform, Wavelet Transform); and (iii) classification by means of machine learning methods. The first promising results from the
classification of AD, MCI, and control samples are published on the
Computational Intelligence and Data mining of the IEEE Symposium Series of
Artificial Intelligence 2014 (CIDM of SSCI 2014).
This work is performed in collaboration with Dr. Maria Cristina De Cola from the IRCCS Centro Neurolesi Bonino Pulejo in Messina (Sicily).
Multiple solution extraction for classification
Biological systems usually offer problems that may have different alternative solutions; in many settings, we are interested in knowing if this is the case,
by counting the number of equivalent solutions, and by listing and comparing them. We call such a type of problem a Multiple-Solutions Problem (MSP). The aim
of this project is to design and test classification algorithms that are able to explore the solution space and to identify a large portion of the set of equivalent interesting
solutions. First, we approach a specific sub problem where we consider features that are consequent in a portion of the solution space according to a certain order,
and we aim to find the smallest subset of those adjacent features within this segment. Identifying small subset of adjacent features able to separate classes of
samples can be really useful in the genome analysis framework, where the cost of sequencing for the whole nucleotides sequences can be really high.
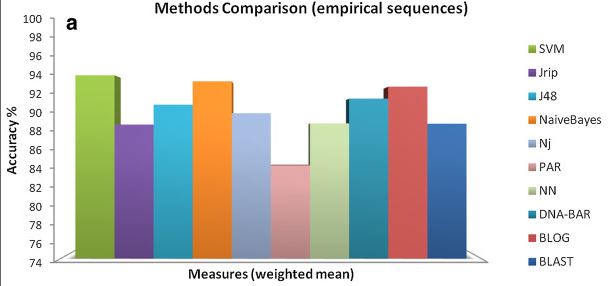
DNA Barcode sequences classification by means of supervised learning techniques
Species classification with DNA Barcode sequences (specific fragments coming from short portions of DNA such as mitochondrial,
nuclear, and plastid sequences) has been proven effective on different organisms.

The DNA Barcodes sequences classification problem may be approached as a supervised machine learning problem in the following way:
given a reference library composed of DNA Barcode specimen sequences of known species and a collection of unknown DNA Barcode sequences
(query set) recognize the latter into the species that are present in the library. Classifier families are tested on synthetic and empirical
datasets belonging to the animal, fungus, and plant kingdoms. In particular, the function-based method Support Vector Machines (SVM),
the rule-based BLOG and RIPPER, the decision tree C4.5, and the Naive Bayes method are considered.
The results are published on several international peer reviewed journals.
Next generation sequencing reads comparison with an alignment free distance
We develop a method to evaluate the similarity between large number of short DNA fragments (reads) extracted from
Next generation sequencing (NGS) machines. This method does not rely on the alignment of the reads and it is based on the
distance between the frequencies of their substrings of fixed dimensions (k-mers). We compare this alignment-free distance
with the similarity measures derived from two alignment methods: Needleman-Wunsch and Blast. We then verify how the
alignment-free and the alignment-based distances reproduce this ideal distance. We exhibit experimental evidence
(through the tests on Saccharomyces cerevisiae, Escherichia coli, and Homo sapiens) that our alignment-free distance
is a potentially useful read-to-read distance measure. Moreover, performing better than the more time consuming distances
based on alignment, our read-to-read distance measure may provide a significant speed-up in several processes based on
NGS sequencing (e.g., DNA reads classification, DNA assembly). The results are published on BMC Research Notes.
Feature selection applied to biological data
When analyzing a huge number of data, a feature selection to reduce the dimension of large data has to be the initial step of the knowledge extraction process,
in order to extract compact and relevant information to aid the classification procedure. Integer programs to model the Feature Selection problem have been adopted,
designing even an ad-hoc randomized solution strategy that obtains comparable performances with respect to other well-known heuristics and achieves good results in a
reasonable time with respect to an Optimal solver that would not accomplish the task. This project is supervised by Dr. Giovanni Felici.